
FDA Oncologic Drug Advisory Committee: October 5th, 2023
Learn about FDA's Oncologic Drug Advisory Committee (ODAC), its role in reviewing oncology drugs, and insights on the October 2023 sotorasib meeting.


Welcome to a learning overview of the FDA’s Oncologic Drug Advisory Committee, ODAC, which advises the FDA and new oncology medications. Per the FDA, the committee, “…reviews and evaluates data concerning the safety and effectiveness of marketed and investigational human drug products for use in the treatment of cancer and makes appropriate recommendations to the Commissioner of Food and Drugs.”1 In short, ODAC’s recommendations help the FDA ensure that new drugs are safe and effective for treating cancer.
The committee consists of experts in general oncology, pediatric oncology, hematologic oncology, immunologic oncology, biostatistics, and related healthcare professions. Throughout each year, meetings are held with sponsors to discuss safety and efficacy data. A 13-member voting committee then votes on a product or products’ use for the treatment of a specific oncology indication.
Today’s article focuses on a previous ODAC. I chose to cover this particular meeting simply because I was interested in the data discussed throughout. As a result, I want to provide a discussion on it here!
Introduction
On October 5th, 2023, an ODAC meeting occurred to discuss data from study CodeBreak-200 and sotorasib for the proposed treatment of adult patients with KRAS G12C mutated locally advanced or metastatic non-small cell lung cancer, NSCLC, who have had at least one prior systemic therapy.
Please see the following October 5th, 2023 ODAC Meeting webpage for publicly available meeting materials, specifically the briefing documents, from the FDA and sponsor company. The briefing documents include discussions and analyses from both the FDA and sponsor company’s perspective and comments or concerns regarding the safety and efficacy data of a product and/or the clinical trial.
Background Information:
Sotorasib is a first-in-class RAS GTPase inhibitor. In May 2021, it was granted accelerated approval for the treatment of adult patients with KRAS G12C mutated locally advanced or metastatic non-small cell lung cancer who have had at least one prior systemic therapy based on CodeBreak-100. Following the accelerated approval, the sponsor was required to conduct a confirmatory trial to verify and describe sotorasib’s clinical benefit. This brings us to CodeBreak-200.
Study Overview:
CodeBreak-200 is a phase III, randomized, open-label confirmatory clinical trial comparing single-agent oral sotorasib vs single-agent IV docetaxel. Dosing regimens include sotorasib 960mg daily and docetaxel 75mg/m2 every three weeks. Key eligibility criteria include the presence of KRAS G12C mutation, advanced or metastatic NSCLC, and progression or disease recurrence on or after receiving at least one prior systemic therapy for locally advanced and unresectable or metastatic disease. Prior therapy must include a platinum-based doublet chemotherapy and checkpoint inhibitor, given as one line of therapy or individual lines of therapy unless the patient has a medical contraindication to one of the required therapies.
Radiographic tumor assessments were completed at the time of screening every 6 weeks through week 49, then every 9 weeks moving forward. Patients received treatment until one of the following: independent central confirmation of disease progression, treatment intolerance leading to discontinuation, initiation of another anticancer therapy, or withdrawal of consent. Per study protocol, patients could crossover from docetaxel to sotorasib after investigator determination of radiologic progression.

ODAC Question:
Each ODAC contains one or more questions that the voting panel discusses and votes on following the presentation of data and arguments from both the FDA and the sponsor company. The question for this particular ODAC is, “Can PFS results of CodeBreak-200 be reliably interpreted?” Important note: The FDA stated that it is not asking about the totality of evidence and benefit/risk, the conversion of accelerated approval to traditional approval, nor whether or not sotorasib should be removed from the treatment market.
FDA Concerns:
The FDA cited six concerns with the trial and the trial results.
- Marginal PFS Improvement
- Asymmetric Early Dropout
- Early Crossover
- Investigator Assessment Bias
- Potential Imaging Charter Violation
- Lack of OS Benefit
Discussion of Each Concern:
1. Marginal PFS Improvement
The primary endpoint in CodeBreak-200 is progression-free survival, or PFS. PFS is defined as the time when a patient is treated to the first documentation of objective disease progression or death by any cause.2 The study’s secondary endpoint is overall survival, or OS. OS is defined as the time from randomization to death.3 For PFS, disease progression was based on blinded independent central review, BICR, of disease response per Response Evaluation Criteria in Solid Tumors version 1.1, or RECIST v1.1. Table 1 below includes data for both endpoints at the time of the ODAC meeting.
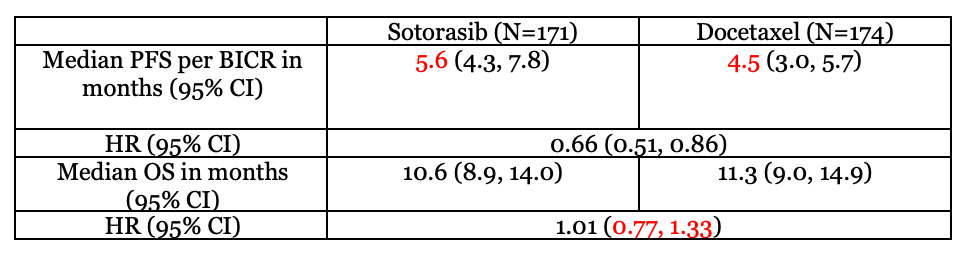
In the ODAC meeting, the FDA noted that PFS as an endpoint is “subjective” in nature and may lead to variation in outcome assessments across different assessors. The FDA also noted that to combat subjectivity, the magnitude of treatment effect on PFS must be large enough to overcome any potential variability in its assessment.
Table 1 shows a statistically signification improvement in median PFS of about 5 weeks. The FDA cited a concern because the study involved a tumor assessment screening schedule of every 6 weeks. This means that the exact date of disease progression is unknown and can be assumed to occur at any point in the six-week imaging interval. With the trial being open-label, the FDA cited a concern for additional bias in tumor assessments. The primary analysis also found no difference in median OS due to its hazard ratio in this analysis crossing one. Overall, the FDA expressed concerns about the uncertainty in the clinical significance of a marginal PFS improvement as well as a lack of OS benefit between sotorasib and docetaxel.
2. Asymmetric Early Dropout
A patient is deemed an early dropout after being randomized to a study arm and deciding to no longer participate in the study before receiving at least one dose of either medication. The sotorasib arm experienced 2 early dropouts, and the docetaxel arm experienced 23 early dropouts. The FDA cited a concern for potential patient preference for sotorasib. The FDA also noted this as a potential loss of randomization due to a loss of balance from early dropouts occurring predominantly on one study arm compared to the other.
The early dropout patients were also censored, which means they had minimal information to implement into the data. This is a potential source of bias that may affect treatment estimations, considering that it is unknown whether or not these patients would have had better or worse outcomes if they stayed in the trial. Overall, the asymmetric early dropouts introduce potential bias favoring sotorasib. Censoring dropout patients leads to a potential overestimation of PFS treatment effect if the dropout patients, mostly on the docetaxel arm, would have had better outcomes.
3. Early Crossover
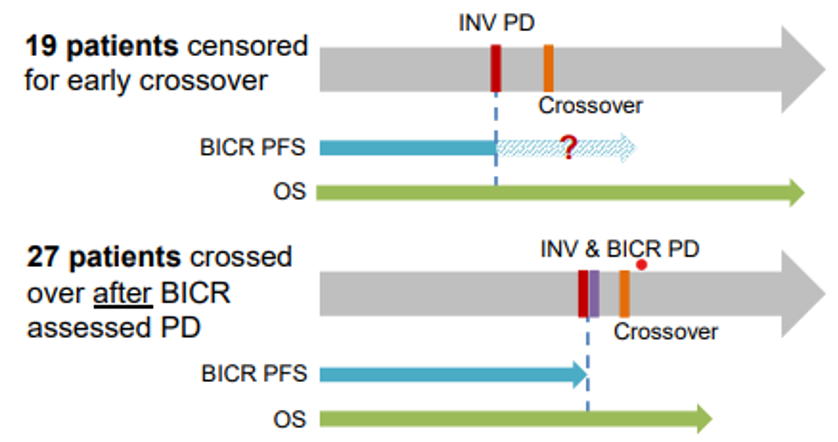
As seen in Figure 1, patients are deemed early crossovers when they transitioned from docetaxel to sotorasib based on investigator-assessed progressive disease, PD, and before BICR-assessed PD. 19 patients experienced early crossover, and all of them were censored at their last BICR by investigator assessment. The FDA cited a concern because the censoring leads to incomplete information for PFS per BICR, the primary endpoint of CodeBreak-200.
27 patients experienced crossover after BICR-assessed PD and started on new anti-cancer therapy. The FDA cited a concern because the new therapy may confound BICR assessments of disease progression. In addition, the sponsor completed an analysis of PFS by BICR for early crossover patients and patients who experienced crossover after both investigator and BICR assessment. At the time of the ODAC meeting, the median post-progression survival for early crossover patients was 18.9 months; the median post-progression survival for patients who experienced crossover after both investigator and BICR assessment was 11.8 months. The FDA then conducted its own exploratory comparison of survival and cited a concern of its analysis indicating that early crossover patients may be healthier than those who crossed over after BICR-assessment of PD, conflating the sponsor’s analysis of median post-progression survival.
4. Investigator Assessment Bias
The FDA analyzed if a difference existed when the investigators determined PD compared to when the BICR determined PD. Within the docetaxel arm, the FDA found a larger number of early determinations of PD for the investigator assessment compared to BICR assessment. Within the sotorasib arm, the FDA found a larger number of later determinations of PD for the investigator assessment compared to BICR assessment. The FDA noted a concern for potential bias favoring the sotorasib arm because its findings indicate that the investigators may be more likely to discontinue docetaxel earlier compared to sotorasib. The findings also indicate that the investigators may be more likely to keep patients on sotorasib longer than docetaxel.
5. Potential Imaging Charter Violation
The imaging charter pertains to a protocol the imaging vendor has when conducting reads on radiographic scans for tumor size and disease progression. Prior to the ODAC meeting occurring, the sponsor observed a higher-than-expected variability between confirmation of disease progression between the procedure and BICR assessment.
The sponsor then contacted its imaging vendor and ultimately conducted a BICR re-read of a select number of patients. This re-read in the initial PFS interim analysis changed from not significant to now being statistically significant. The FDA considered this re-read to be a potential violation of the imaging charter, which states that vendor assessments are not subject to input from the sponsor. Through an Ad-Hoc meeting prior to the ODAC meeting, the FDA advised against submission of its marketing application with data involving the potential imaging charter violation and recommended a global re-read of all patient scans.
6. Lack of Overall Survival Benefit
The median overall survival, OS, for both treatment arms are included in Table 1. The median OS for sotorasib is 10.6 months (95% CI: 8.9, 14.0). The median OS for docetaxel is 11.3 months (95% CI: 9.0, 14.9). The hazard ratio in the primary analysis is 1.01 (95% CI: 0.77, 1.33). As seen with the hazard ratio crossing one, the results are not statistically significant. At the time of the ODAC meeting, the FDA cited concern about a lack of improvement in OS with sotorasib treatment compared to docetaxel.
The ODAC committee ultimately voted 10-2 that the primary endpoint, PFS by BICR, in Codebreak-200 could not be reliably interpreted. It is important to remember that the FDA does not intend to remove sotorasib from the treatment market, and multiple regulatory pathways are available to keep sotorasib approved while the medication is studied further. For further coverage of this ODAC meeting, please see the following Oncology and Fierce Pharma.
References:
- U.S. Food and Drug Administration. Oncologic Drugs Advisory Committee.
- Faiman B. The Importance of Endpoints in Oncology Clinical Trials. J Adv Pract Oncol. 2023;14(6):466-467.
- Delgado A, Guddati AK. Clinical Endpoints in Oncology - A Primer. Am J Cancer Res. 2021;11(4):1121-1131.
*Information presented on RxTeach does not represent the opinion of any specific company, organization, or team other than the authors themselves. No patient-provider relationship is created.